Genoma e Variações Genéticas dos Coronavírus Destaque
- Escrito por Portal Ciencia.ao
- tamanho da fonte diminuir o tamanho da fonte aumentar o tamanho da fonte

Fonte: https://www.michaeljfox.org/news/cost-genome-sequencing-falling-low-cost-alone-wonaeutmt-translate-genes-treatments
António A. N. de Alcochete
Departamento de Biologia, Faculdade de Ciências, Universidade Agostinho Neto;
Email: Este endereço de email está protegido contra piratas. Necessita ativar o JavaScript para o visualizar.; Telef: (+244)924440694
INTRODUÇÃO
O conhecimento da genética, com recurso a tecnologias inovadoras e metodologias que possibilitam a análise e comparação de multiplos genomas dos vírus, vectores e hospedeiros, em função do tempo e do espaço é deveras bastante informativo pois permite entender a origem, a evolução e a dispersão geográfica da doença.
O termo genoma, introduzido em 1920 por Hans Winkler, foi utilizado como referência de toda a informação hereditária de um organismo que está codificada em seu DNA ou RNA, ou simplesmente, de uma sequência de DNA completa de um conjunto de cromossomos. Actualmente, o conceito de genoma compreende a informação necessária para construir, manter e conhecer a história evolutiva de um organismo (Cristescu, 2019).
1. GENOMA DOS CORONAVÍRUS
O genoma dos coronavírus é uma molécula de ARN de fita simples, sentido positivo, cujo tamanho varia entre 27 a 32 kpb e contém pelo menos seis “Open Reading Frames” (ORFs). As primeiras ORF (ORF1a/b), localizadas no extremo 5´, ocupam cerca de dois terços do genoma e codificam a poliproteína 1a,b (pp1a, pp1b). As restantes ORFs estão localizadas no extremo 3´e codificam, pelo menos, quatro proteínas estruturais: a gliproteína espiculada da cápside/envelope (S), responsável pelo reconhecimento dos receptores da celúla hospedeira; as proteínas de membrana (M), responsável pela forma da cápside/envelope; as proteínas da cápside/envelope (E), responsável pela montagem e liberação dos vírus; as proteínas nucleocapsídicas (N) envolvidas no empacotamento do genoma que desempenham um papel na patogenicidade como inibidor do interferon (IFN). Existem também proteínas estruturais e acessórias espécie-especificas, tais como as proteínas HE, 3a/b e 4a/b (Alanagreh et al., 2020).
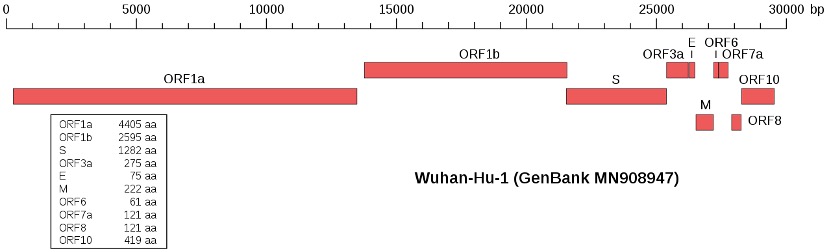
Fig 1 – Genoma do SARS-Cov-2 (Fonte: Wu et al., 2020a).
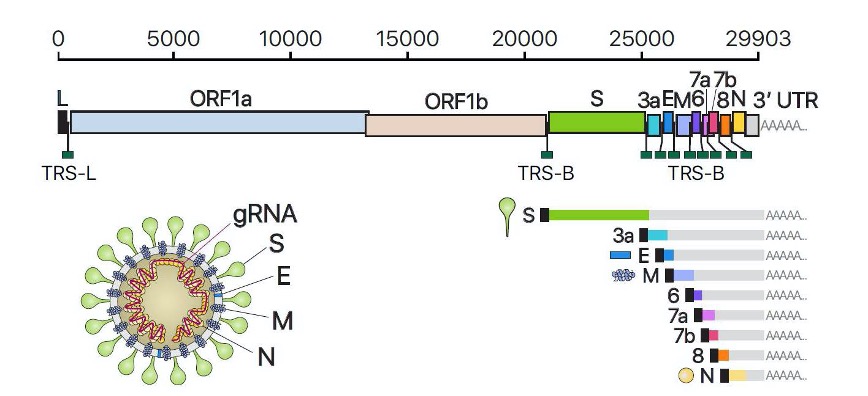
Fig 2 – Genoma do SARS-Cov-2 mostrando os ARN genómicos e subgenómicos ((S, E, M, N, 3a, 6, 7a, 7b, 8) (Fonte: Fernandez-Rua, 2020)
2. VARIAÇÕES GENÉTICAS
Apesar do pequeno tamanho do genoma dos coronavírus, o processo evolutivo adapatativo em diferentes ambientes hospedeiros e a grande dispersão geográfica permite registar alterações genéticas estruturais e funcionais. Estudos de análise genética comparativa têm mostrado que os genomas dos coronavírus conservam entre 50% a 95% de semelhança. O genoma do SARS-CoV-2, que infecta o ser humano, parece conter até 15 genes muito semelhante ao SARS-CoV encontrado em Manis javanica (Pangolin) e nos morcegos, especialmente com o vírus Beta-CoV encontrado em morcegos, em 96,2%, e com Bat-CoV-RatTG13, em 79,2% com SARS-CoV (Zhu et al., 2020).
Embora seja notável a similaridade dos genomas, a subunidade da proteína S do vírus do Pangolim mostrou maior similaridade com SARS-CoV-2 do que com SARS-CoV e com o Bat-RaTG13.
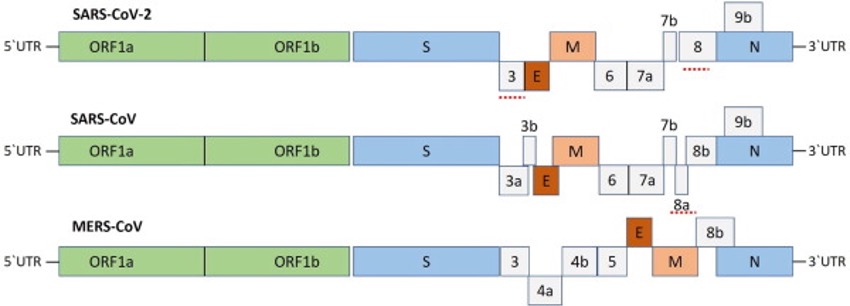
Fig 3 – Similaridade genética e variações dos genomas de SARS-Cov-1, SARS-Cov e MERS-Cov (Fonte: Shereen et al., 2020)
As alterações genéticas são também refletidas pela elevada taxa de mutação mostrada pelos coronavírus. Foster et al. (2020) aplicaram um algoritmo matemático numa análise filogenética de 160 genomas de pacientes humanos, tendo identificado três variantes de SARS-Cov-2 (A, B e C), sendo que as variantes A e C são encontradas na Europa e América enquanto a variante B é encontrada na Ásia. Entretanto, deve-se realçar que a vaiante A, que é o genoma original do vírus identificado em Wuhan, não foi a variante predominante na cidade.
Xiaolu et al. (2020) analisaram 103 genomas de SARS-Cov-2, tendo identificado duas estirpes desse vírus, designados L e S, diferenciados por dois polimorfismos de nucleótido simples ou polimosfismos de nocleótido único (SNPs). A esitrpe L foi identificado como sendo mais prevalente na amostra do que a estirpe S. A falta de clareza sobre a implicação dessas alterações evolutivas na etiologia da doença sugerem a necessidade de estudos mais aprofundados do vírus. A análise do gene S viral que interage como receptor da célula hospedeira indicidou a ocorrência de recombinação genética (Wu et al., 2020b).
Zhang et al. (2020) sugeriram a ocorrência de mutações como resultado da classificação de 27 genomas em seis grupos genéticos, apesar da alta similaridade, num estudo de 27 pacientes de três cidades da China (Wuhan, Zejiang e Guangdong) e da Tailândia, todos com contactos a partir de Wuhan. Estes especialistas verificam que o grupo genético mais basal era de Guangdong e que o grupo do novo coronavírus apresentava 380 substituições de aminoácidos.
Laarmarti et al. (2020) analizaram 3067 genomas de SARS-CoV-2 provenientes de 59 países, com recurso a análise genomica comparativa, por meio de perfis das mutações e comparação das frequências, bem como a monitorização da sua geografia. Este grupo de cientistas identificou 716 mutações, sendo 457 mutações com efeito sem-sinónimo, 39 mutações recorrentes de efeito não-sinónimo, incluindo 10 mutações “hot-spot” com prevalência superior a 0,10 distribuídas em seis genes do SARS-Cov-2. O estudo mostrou genótipos específicos as localidades e a ocorrência simultânea de mutações devido a presença de vários haplótipos, sugerindo a acção de um mecanismo de co-acumulação de mutações e um agrupamento dos vírus em 3 sub-grupos.
Coppe et al. (2020) relataram 2334 mutações não sinónimas após análise de sequências de SARS-CoV-2 obtidas do CoV_GLUE5 37 (http://cov-glue.cvr.gla.ac.uk/: 9,028 available sequences (‘low coverage’ excluded), incluindo 4973 sequências de pacientes europeus. As duas principais mutações (S-D614G & nsp12-P323L) que divergem do virus SARS-CoV-2, Referência NCBI (NC_045512), são verificadas em todos os continents, com apenas 3 casos na Ásia. Foram identificadas mutações D614G na proteína S (encontrada em 2342 amostras), que determina o Grupo G, e co-evolui com a mutação P323L na proteína nsp12 (encontrada em 2318 amostras); a mutação ORF8-L84S (terceira mutação mais frequente), que determina o Grupo S; a mutação substituição do aminoácido L84S que co-evolui com outras três mutações: 55 nsp4-F308Y, ORF3a-G196V e N-S197L. Estas últimas três mutações, juntamente com as mutações S197L e a substituição de P13L na proteína N são pouco frequentes; a quarta mutação mais frequente é ORF3a-Q57H encontrada em 734 sequências; seguem-se as mutações N-R203K e N-G204R) encontradas na Europa; e, por último, referimos as mutações nsp6-L37F e ORF3a-G251V, correspondendo ao Grupo V.
Dorp e Balloux (2020) concluiram que as sequências genómicas estudadas partilham um ancestral comum que corresponde ao periodo que o SARS-CoV-2 infectou, pela primeira vez, o homem. Estes autores identificaram regiões do genoma que não variaram e 198 mutações recorrentes, sendo que aproximadamente 80% produziram alterações não-sinónimas na proteína S; mais de 15% de mutações recorrentes nas regiões Nsp6, Nsp11 e Nsp13 da ORF1ab e na proteína S, indicando uma evolução convergente de particular interesse no processo adaptativo do SARS-Cov-2 ao homem.
Eskier et al., (2020) ao estudar os efeitos das mutações ARN-dependente ARN polymerase (RdRp), em especial a mutação 4408C>, sobre a taxa de mutação e dispersão do vírus, concluíram que a mutação 14408C>T aumenta a taxa de mutação, enquanto que a mutação 15324C>T da RdRp, tem efeito contrário, sugerindo que a mutação 14408C>T pode ter contribuído para a dominância das suas co-mutações em diferentes regiões. Vankadari (2020), numa análise de sequências genómicas virais completas de 12 países diferentes, identificou 47 SNPs com impacto na virulência e resposta contra antivirais, sendo que as proteinas Nsp1, RdRp da gliproteina “pico” e a região ORF8 mutaram no periodo de 3 meses de transmissão humana.
Yang et all., (2020) identificaram seis agrupamentos filogenéticos com preferência geográfica, na análise de 1932 sequências genómicas completas do SARS-CoV-2. Estes autores acreditam que variações nucleótidicas simples (SNVs) em genomas estão na base dos resultados, bem como aportam contributos para a detecção, tratamento clinico, desenho de drogas e desenvolvimento de vacinas contra o virus.
Segundo Sen et al. (2020), o genoma do coronavírus possui seis a sete principais ORFs no sentido 5-3': as ORF1a e 1b que compreendem dois terços do genoma e codificam as poliproteínas não estruturais e quatro ORFs, à jusante, que codificam proteínas estruturais: a proteína de pico (S), a proteína de envelope (E), a proteína de membrana (M) e a proteína de nucleocapsídeo (N). Alguns coronavírus têm um gene de hemaglutinina-esterase (HE) entre ORF1b e S. Além dos genes conservados em coronavírus, o genoma de SARS-CoV contém vários genes acessórios específicos, incluindo ORF3a, 3b, ORF6, ORF7a, 7b, ORF8a, 8b e 9b.
3. CONCLUSÕES
Apesar do genoma do coronavírus ser pequeno e compreender uma molécula de ARN, de fita simples, o sentido positivo da molécula permite-lhe a sua rápida tradução imediatamente após a infecção das células hospedeiras, produzindo assim as proteínas necessárias para a sua replicação. Por outro lado a alta taxa de infecção, a alta especifidade da proteina “pico” (S) para com o receptor das células hospedeiras e a larga dispersão geográfica são qualidades inerentes a capacidade de mutação estrutural e funcional dos vírus. A consulta mostra o grande número de mutações registadas nos genomas dos virus, com diferentes graus de importância e têm sido classificadas como recorrentes (frequentes no espaço e no tempo), não-sinónimas (pontuais, ou seja, envolvem a substituição de um único nucleótido), tendo sido identificadas regiões “hot spot” (regiões onde as mutações ocorrem a uma frequência mais alta).
4. REFERÊNCIAS BIBLIOGRÁFICAS
Cristescu, Melania E. The concept of genome after one century of usage. Genome 62: iii–v (2019) dx.doi.org/10.1139/gen-2019-0129
Doğa Eskier, Gökhan Karakülah, Aslı Suner, Yavuz Oktay (2020) RdRp mutations are associated with SARS-CoV-2 genome evolution. bioRxiv preprint doi: https://doi.org/10.1101/2020.05.20.104885
Fernandez-Rua, José M (2020) Nuevo mapa genético del SARS-Cov-2. Biotech magazine & News, https://biotechmagazineandnews.com/nuevo-mapa-genetico-del-sars-cov-2/.
Hsin-Chou Yang, Chun-houh Chen, Jen-Hung Wang, Hsiao-Chi Liao, Chih-Ting Yang, Chia-Wei Chen, Yin-Chun Lin, Chiun-How Kao, and James C. Liao (2020) Genomic, geographic and temporal distributions of SARS-CoV-2 mutations. bioRxiv preprint doi: https://doi.org/10.1101/2020.04.22.055863
Lucyvan Dorp and François Balloux (2020) Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infection, Genetics and Evolution, Volume 83, September 2020, 104351. https://doi.org/10.1016/j.meegid.2020.104351
Shereen, M.A., Khan, S., Kazmi, A., Bashir, N. and Siddique, R. (2020) COVID-19 infection: Origin, transmission, and characteristics of human Coronaviruses. Journal of Advanced Research 24(2020):91-98.
Sen S, Anand KB, Karade S, Gupta RM. Coronaviruses: origin and evolution. Medical Journal Armed Forces India. 2020 Apr 27;76(2):136–41. doi: 10.1016/j.mjafi.2020.04.008. Epub ahead of print. PMID: 32341622; PMCID: PMC7183968.
Vankadari, N. (2020) Overwhelming mutations or SNPs of SARS-CoV-2: A point of caution. Gene. 2020 May 20;752:144792. doi: 10.1016/j.gene.2020.144792. Epub ahead of print. PMID: 32445924; PMCID: PMC7239005.
Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., and Song, Z. G., et al . (2020a) A new coronavirus associated with human respiratory disease in China. Nature [Epub ahead of print]. https://doi.org/10.1038/s41586-020-2008-3
Wu, A., Peng, Y., Huang, B., Ding, X., Wang, X., Niu, P., et al . (2020b) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host & Microbe [Epub ahead of print]: 1931– 3128.
Xiaolu Tang, Changcheng Wu, Xiang Li, Yuhe Song, Xinmin Yao, Xinkai Wu, Yuange Duan, Hong Zhang, Yirong Wang, Zhaohui Qian, Jie Cui, Jian Lu (2020) On the origin and continuing evolution of SARS-CoV-2 . National Science Review. https://doi.org/10.1093/nsr/nwaa036
Zhang, L., Shen, F. M., Chen, F., and Lin, Z. (2020a) Origin and evolution of the 2019 novel coronavirus. Clinical Infectious Diseases [Epub ahead of print]. pii: ciaa112.
Zhu N, Zhang D, Wang W, et al. (fevereiro de 2020). «A Novel Coronavirus from Patients with Pneumonia in China, 2019». The New England Journal of Medicine. 382 (8): 727–733. PMID 31978945. doi:10.1056/NEJMoa2001017
Últimas de Portal Ciencia.ao
- RECRUTAMENTO: PDCT-MESCTI lança Concurso para a contratação de Secretária(o)
- PDCT-MESCTI: AVISO DE SOLICITAÇÃO DE COTAÇÃO - DESENVOLVIMENTO CONCEPTUAL E CRIATIVO DA LOGOMARCA E DE ESCULTURAS DO PCTL
- PDCT-MESCTI: Anúncio de Assinatura de Adenda N°2 do Contrato: "Projecto, Fiscalização da Construção e Plano de Negócio do Parque de Ciência e Tecnologia de Luanda
- PDCT-MESCTI realizou visita de acompanhamento aos seus bolseiros na Universidade de São Paulo e visitou o Instituto Butantan
- Acompanhe a Cerimónia de Premiação (13/12/2024)- Prémio Nacional de Ciência e Inovação 2024